









Mündəricat
Hemofiliya
Bu nədir ?
Hemofiliya, qanın laxtalanmasını maneə törədən, yaralandıqda və bəzən yaralanmadan qeyri -adi uzun qanaxmalara səbəb olan genetik hemorragik bir xəstəlikdir. Bir qan laxtasının qanamasını dayandıracaq qədər güclü bir şəkildə meydana gəlməsini maneə törədən pıhtılaşma zülallarının olmamasına və ya olmamasına səbəb olan bir genetik mutasiyadan qaynaqlanır.
Hemofiliya oğlanların bir xəstəliyi olaraq qəbul edilir və hər 1 oğlan uşağından 5000 -i təsirlənir. Ancaq qızlar genetik mutasiyanı daşımaqla xəstəliyin kiçik bir formasını inkişaf etdirə bilərlər. Ümumi əhalidə hər 1 nəfərdən 12 -də hemofiliya var. (000) Xəstəliyin iki əsas növü vardır: hemofiliya A və B. Hemofiliya A -nın yayılması hemofiliya B -dən daha yüksəkdir (1/1 kişi 6 /000 -ə qarşı). (1).
Hemofiliya hələ bir neçə onilliklər əvvəl uşaqlıqdan və gənclikdən bəri çox zəiflədən və ölümcül bir xəstəlik idi. Bu gün təsirli, lakin məhdudlaşdırıcı müalicələr hemofiliyalı insanlarda qanaxmanın qarşısını almağa, bədən xəsarəti və əlilliyi məhdudlaşdırmağa imkan verir.
Belirtiler
uşaq. Uzun müddətli qanaxma bir zədə və ya kiçik bir travmadan sonra baş verir. Xəstəliyin ağır formalarında kortəbii ola bilərlər (buna görə də travma olmadıqda müdaxilə edə bilərlər). Qanama daxili və ya xarici ola bilər. Diqqət yetirin ki, hemofiliyalı bir insanda qanaxma daha şiddətli deyil, müddəti daha uzun olur. Əzələlərdə (çürüklərdə) və oynaqlarda (hemartroz), əsasən topuqlarda, dizlərdə və kalçalarda qanaxma zaman keçdikcə iflicə səbəb ola biləcək sərtliyə və əlil deformasiyalara səbəb ola bilər.
Xəstəlik, qanda laxtalanma faktorlarının miqdarı aşağı olduqda daha da şiddətlənir (1):
- Şiddətli forma: spontan və tez -tez qanaxmalar (50% hallarda);
- Orta forma: kiçik zədələrdən sonra anormal uzun qanaxmalar və nadir kortəbii qanaxmalar (10-20% hallarda);
- Kiçik forma: anormal uzun qanaxmalar, lakin spontan qanaxmaların olmaması (30-40% hallarda).
Xəstəliyin mənşəyi
Qanda laxtalanma faktorları adlanan zülallar var ki, bu da qan laxtasının əmələ gəlməsinə imkan verir və buna görə də qanaxmanı dayandırır. Genetik mutasiyalar bu zülalların istehsalına mane olur. Hemofiliya A və B ilə əlaqəli simptomlar çox oxşardırsa, xəstəliyin bu iki növü yenə də fərqli bir genetik mənşəyə malikdir: hemofiliya A, laxtalanma faktorunu VIII və l hemofiliya B kodlayan F8 genindəki (Xq28) mutasiyalardan qaynaqlanır. laxtalanma faktorunu IX kodlayan F9 genində (Xq27).
Risk faktorları
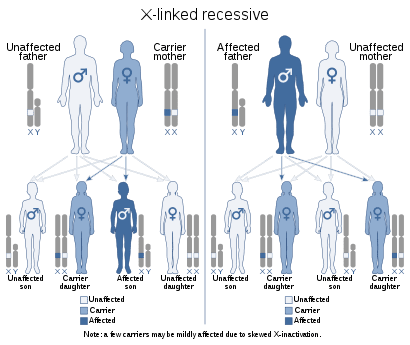
Hemofiliyaya X xromosomunda yerləşən genlər səbəb olur. "X ilə əlaqəli resesif irs" olaraq bilinən bir genetik xəstəlikdir. Bu, xəstə bir insanın dəyişdirilmiş geni yalnız qızlarına və oğlanlarına 50% risklə ötürə biləcək qızlarına ötürəcəyini nəzərdə tutur. Nəticədə xəstəlik demək olar ki, yalnız kişilərə təsir edir, ancaq qadınlar daşıyıcıdır. Hemofiliyanın təxminən 70% -nin ailə tarixi var. (1) (3)
Qarşısının alınması və müalicəsi
Müalicələr artıq qanaxmanın qarşısını almağa və cilovlamağa imkan verir. Onlar antihemofil amilin venadaxili tətbiqindən ibarətdir: hemofiliyalılar üçün VIII faktoru və hemofiliyalılar üçün IX faktoru B. Bu antihemofil dərmanlar qandan (plazma mənşəli) alınan məhsullardan və ya mühəndislik tərəfindən istehsal olunur. genetika (rekombinantlar). Onlar qanaxmanın qarşısını almaq üçün və ya qanaxma hadisəsindən sonra müntəzəm və sistematik inyeksiyalarla aparılır. Fizioterapiya hemofiliyalı insanlara əzələlərin elastikliyini və təkrar qanaxmalara məruz qalmış oynaqların hərəkətliliyini qorumağa imkan verir.